
Share
Follow Us
HEPATOLOGY, January 1998, p. 289-291, Vol. 27, No. 1
Editorial
Mutations in the Hemochromatosis Gene, Porphyria Cutanea Tarda, and Iron Overload
It has long been thought that the inheritance of one or more human leukocyte antigen (HLA)-linked hemochromatic genes maycontribute to iron overload in diseases other than genetic hemochromatosis(GH).1 Porphyria cutanea tarda (PCT) has always been a primecandidate for this association. Patients with this disorder presentwith fragile skin and subepidermal bullae and often show someevidence of hepatocyte damage, although cirrhosis is unusual.2Mild to moderate iron overload is present in 60% to 70% of cases. 3,4 PCT seems to be provoked in most patients by alcohol, estrogens,viruses (notably hepatitis C virus and human immunodeficiencyvirus), or a combination of these factors. It is rarely associatedwith clinically overt GH. 2,5
The underlying metabolic abnormality is decreased activity of uroporphyrinogen decarboxylase, one of the enzymes of the hemebiosynthesis, in the liver. Clinical and experimental studiessuggest that this enzyme is reversibly inactivated by an iron-dependentprocess.6 Depletion of hepatic iron stores by venesection,or by other means, leads to clinical remission and reversal ofthe metabolic defect, even in those without iron overload.2In animal models of PCT, the inactivation process is both dependenton iron and accelerated by iron overload. 7,8
Although development of PCT in response to hepatocyte damage by these agents is believed to be determined by inherited factors,few genes that contribute to this predisposition have been identified.Most patients have the sporadic (type I) form of PCT in whichthe enzyme defect is restricted to the liver and in which causativemutations at the uroporphyrinogen decarboxylase locus have beenexcluded.9 Even in the 20% or so of patients from familiesshowing autosomal dominant inheritance of half-normal enzyme activityin all tissues (familial or type II PCT), further inactivationof the enzyme in the liver seems necessary for clinical expression. 6,9,10
About 75% of patients with genetic hemochromatosis carry the HLA-A3 allele compared with 25% of controls. Some studies ofthe frequency in PCT of this and other alleles defining the ancestralhemochromatosis haplotype (HLA-A3, D6S265-1, D6S105-8, D6S1260-4)have shown an association, 3,4,11,12 whereas others havenot.13-16 In 1996 Feder et al.17 cloned a strong candidate forthe hemochromatosis HFE gene which was located 4.5 Megabases telomericto the HLA-A locus and encoded an HLA-class Ib protein. They foundthat about 85% of chromosomes from patients with GH carry a pointmutation in the HFE gene which replaces cysteine at amino acidposition 282 with tyrosine (C282Y). A second mutation which replaceshistamine 63 by aspartic acid (H63D) is common in the population(gene frequency 0.15); however, by itself it is not associatedwith iron overload. The wild-type and H63D proteins are expressedon the cell surface and bind ![]() 2 microglobulin (like most classI-HLA proteins) but the mutant C282Y protein neither reaches thecell surface nor binds
2 microglobulin (like most classI-HLA proteins) but the mutant C282Y protein neither reaches thecell surface nor binds ![]() 2 microglobulin.18 The two mutationsare in complete linkage disequilibrium. Compound heterozygotes,which comprise about 1% of the general population, may developiron overload and even clinical hemochromatosis.19-21
2 microglobulin.18 The two mutationsare in complete linkage disequilibrium. Compound heterozygotes,which comprise about 1% of the general population, may developiron overload and even clinical hemochromatosis.19-21
The C282Y mutation is responsible for much of the iron overload in populations of European descent. All GH patients in Queensland,2292% in both Brittany23 and the UK,19 about 83% in the USA, 17,21 71% in France generally,24 and 61% in Italy25 are C282Y homozygotes.In parts of Northern Europe, the gene frequency in the generalpopulation approaches 10% but falls to 3% in Greece and to only0.5% in Italy. The mutation seems to be absent from the nativepopulations of Asia, Africa, the Middle East, and the Americas.26
Early in 1997, Roberts et al.27 reported that 44% of 41 British patients with sporadic PCT carried at least one copy ofthe C282Y mutation. This association has since been confirmedin patients with sporadic PCT from the Netherlands,28 the USA,29Australia,30 and, for patients of European descent, South Africa.31It is also present in familial PCT. 28,32
In this issue of HEPATOLOGY, Sampietro et al.33 report that the association between the C282Y mutation and sporadic PCTis not found in Italian patients. The allele frequency in 68 malepatients (1.5%) was not increased in comparison with two controlgroups: 28 students and hospital staff (0.7%), and because hepatitisC virus infection is common in Italian patients with PCT, 50 patientswith chronic hepatitis caused by hepatitis C virus (2%). Thoughthe frequency of this mutation in the general population and inGH is lower than in Britain, this finding is unexpected particularlyas the range of iron stores in Italian patients with PCT doesnot differ from patients elsewhere.3-5 Even more unexpectedly,the frequency of the other mutation in the HFE gene, H63D, wassignificantly increased as it was present on 28.7% of HFE allelesin the PCT group compared with 12.8% and 12% in the two controlgroups. There was no relationship between the presence of thismutation and the degree of iron storage. To our knowledge, thisis the first report of an association of this mutation alone witha disease.
Five of the Italian patients without HFE mutations but with iron overload carried the ancestral hemochromatosis haplotype,one of which was homozygous. The authors point out that theremay be another gene, linked to the ancestral haplotype, that causeshemochromatosis. Patients homozygous for the ancestral haplotypeare reported to accumulate more iron than other patients do withGH in both Queensland34 and Italy.35 In the study by Robertset al.,27 four chromosomes with the ancestral haplotype alsolacked the C282Y mutation. However, in Britain, this mutationis only carried by 50% of chromosomes with the ancestral haplotypeand there may not be any other iron loading genes associated withit.36
Is the frequency of the H63D mutation increased in other populations with PCT? The C282Y mutation has predominated in allother series reported to date.27-32 As the two mutations are incomplete linkage disequilibrium, calculating the frequency ofthe H63D chromosome after eliminating those carrying the C282Ymutation has been advocated. 17,37 This calculation did notreveal any significantly higher frequency in 107 British patientswith sporadic PCT. 27,32 Further studies of HFE mutations inPCT from other countries where the population frequency of theC282Y mutation is lower than in Northern Europe are awaited withinterest.
What conclusions may be drawn from these studies of PCT? The main one is that HFE mutations confer susceptibility to PCT.The mechanism is far from clear. PCT is rare in heterozygotesfor each mutation. Presumably the C282Y mutation promotes ironaccumulation and may, thereby, accelerate the onset of diseasein those who are already predisposed. The relationship betweenthe presence of this mutation and iron storage in PCT has notyet been established. Sampietro et al. suggest that the H63D mutationmay cause a subtle change in iron metabolism caused by the accumulationof toxic iron species which accelerates inactivation of uroporphyrinogendecarboxylase. Hepatitis C virus infection is much more commonin Italian patients with PCT than in British patients with PCT.5The authors hypothesize that hepatitis C virus infection and theH63D mutation might synergize to produce clinically manifest PCT,whereas the C282Y mutation might more efficiently trigger PCTindependently of viral liver disease.
Will there be other diseases in which these mutations play a role? In some populations hemochromatosis is common, and, therefore,it is inevitable that some patients with iron overload secondaryto hematological disorders will also carry a gene for hemochromatosis.1Of two brothers with pyridoxine-responsive sideroblastic anemia,one carried both the C282Y and H63D mutations and had accumulatedmore iron than his brother with a normal genotype.38 More extensiveinvestigations of HFE mutations in such patients are awaited.
A final unexpected finding from these studies has been the high proportion of PCT patients who are C282Y homozygotes (21%in Britain). In countries where this mutation is common, patientswith PCT should be screened for its presence. Homozygotes shouldbe treated by venesection and monitored to ensure that iron doesnot reaccumulate, and their families should be investigated. InGH it is suggested that the transferrin saturation should be keptbelow 55% and that the serum ferritin below 100 µg/L. Similarstandards should apply to PCT.
| George H. Elder, M.D. Department of Medical Biochemistry |
||||||||
| Mark Worwood, Ph.D. Department of Hematology University of Wales College of Medicine Heath Park, Cardiff, UK |
Footnotes
Abbreviations: HLA, human leukocyte antigen; GH genetic hemochromatosis, PCT, porphyria cutanea tarda.
Received November 3, 1997; accepted November 5, 1997.
Address reprint requests to: Professor G H Elder, M.D., Department of Medical Biochemistry, University of Wales College of Medicine, Heath Park, Cardiff CF4 4XN, UK. Fax: 44-1222-744905.
REFERENCES
- Simon M. Secondary iron overload and the haemochromatosis allele. Br J Haematol 1985; 60: 1-5.
- Kappas A, Sassa S, Galbraith RA, et al. The porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Molecular and Metabolic Basis of Inherited Disease. 7th ed. New York, NY: McGraw-Hill, 1995:2103-2159.
- Edwards CQ, Griffen LM, Goldgar DE, et al. HLA-linked hemochromatosis alleles in sporadic porphyria cutanea tarda. Gastroenterology 1989; 97: 972-981.
- Fargion S, Fracanzani AL, Romano R, et al. Genetic haemochromatosis in Italian patients with porphyria cutanea tarda: possible explanation for iron overload. J Hepatol 1996; 24: 564-569.
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis; in press.
- Elder GH, Urquhart AJ, De Salamanca RE, et al. Immunoreactive uroporphyrinogen decarboxylase in the liver in porphyria cutanea tarda. Lancet 1985; 2: 229-233.
- Sweeny GD. Porphyria cutanea tarda, or the uroporphyrinogen decarboxylase deficiency diseases. Clin Biochem 1986; 19: 3-14.
- Constantin D, Francis JE, Akhta RA, et al. Uroporphyria induced by 5-aminolevulinic acid in Ahrd SWR mice. Biochem Pharmacol 1996; 52: 1407-1413.
- Garey JR, Franklin KF, Brown AD, et al. Analysis of uroporphyrinogen decarboxylase complementary DNAs in sporadic porphyria cutanea tarda. Gastroenterology 1993; 105: 165-169.
- Sierseema PD, Rademakers LHP, Cleton MI, et al. The difference in liver pathology between sporadic and familial forms of porphyria cutanea tarda – the role of iron. J Hepatol 1995; 23: 259-267.
- Kuntz BME, Goerz G, Merk H, et al. HLA-A3 and B7 in porphyria cutanea tarda. Tiss Antigens 1984; 24: 67-69.
- Roberts AG, Whatley SD, Nicklin S, et al. The frequency of haemochromatosis-associated alleles is increased in British patients with sporadic porphyria cutanea tarda. HEPATOLOGY 1997; 25: 159-161.
- Santioanni P, de Felice M, Ayala F, et al. A novel association between HLA and disease: porphyria cutanea tarda and DLA-AW32. Dermatologica 1980; 160: 371-375.
- Llorente L, Salamanca RE, Campillo F, et al. HLA and porphyria cutanea tarda. Arch Dermatol Res 1980; 269: 209-210.
- Kostler VE, Gebhardt B, Seebacher C. HLA system and porphyria cutanea tarda. Dtsch Z Verdau Stoffwechselkr 1984; 44: 95-100.
- Beaumont C, Fauchet R, Phung LN, et al. Porphyria cutanea tarda and HLA-linked hemochromatosis: evidence against a systematic association. Gastroenterology 1987; 92: 1833-1838.
- Feder JN, Gnirke A, Thomas A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-408.
- Feder JN, Tsuchihashij Z, Irrinka A, et al. The hemochromatosis founder mutation in HLA-H disrupts
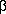 2-microglobulin interaction and cell surface expression. J Biol Chem 1997; 272: 14025-14028.
2-microglobulin interaction and cell surface expression. J Biol Chem 1997; 272: 14025-14028. - The UK Haemochromatosis Consortium. A simple genetic test identifies 90% of UK patients with haemochromatosis. Gut 1997;in press.
- Martinez PA, Biron C, Blanc F, et al. Compound heterozygotes for hemochromatosis gene mutations: may they help to understand the pathophysiology of the disease? Blood Cells Mol Dis 1997; 23: 269-276.
- Beutler, Gelbart T, West C, et al. Mutation analysis in hereditary hemochromatosis. Blood Cells Mol Dis 1996; 22: 187-194.
- Jazwinska EC, Cullen LM, Busfield F, et al. Haemochromatosis and HLA-H. Nat Genet 1996; 14: 249-251.
- Jouanolle AM, Fergelot P, Gandon G, et al. A candidate gene for hemochromatosis: frequency of the C282Y and H63D mutations. Hum Genet 1997; 100: 544-547.
- Borot NN, Roth MP, Malfroy L, et al. Mutations in the MHC class I-like candidate gene for hemochromatosis in French patients. Immunogenetics 1997; 45: 320-324.
- Carella M, D’Ambrosio L, Totaro A, et al. Mutation analysis of of HLA-H gene in Italian hemochromatosis patients. Am I Hum Genet 1997; 60: 828-832.
- Merryweather-Clarke AT, Pointon JJ, Shearman JD, et al. Global prevalence of putative haemochromatosis mutations. J Med Genet 1997; 34: 275-278.
- Roberts AG, Whately SD, Morgan RR, et al. Increased frequency of the haemochromatosis Cys282Tyr mutation in sporadic porphyria cutanea tarda. Lancet 1997; 349: 321-323.
- Santos M, Clevers HC, Marx JJM. Mutations of the hereditary haemochromatosis candidate gene HLA-H in porphyria cutanea tarda. N Engl J Med 1997; 336: 1327-1328.
- Kushner JK. International Symposium on Iron in Biology and Medicine. St. Malo 1997.
- Stuart KA, Busfield F, Jazwinska EC, et al. The C282Y mutation in HFE is common in Australian patients and can act independently of HCV to precipitate porphyria cutanea tarda [Abstract]. HEPATOLOGY 1997; 26: 606A.
- Hift RJ, Corrigall AV, Meissner PN, Kirsch RE. Significance of HLA mutations in South African patients with porphyria cutanea tarda. Acta Haematol 1997;98:(suppl. 1):112.
- Jackson H, Whatley SD, Morgan RR, et al. Haemochromatosis C282Y mutation in porphyria cutanea tarda. International Symposium on Iron in Biology and Medicine, Saint-Malo, 1997 Meeting abstract.
- Sampietro M, Piperno A, Lupica L, Arosio C, Vergani A, Corbetta N, Malosio I, et al. High prevalence of the His63Asp HFE mutation in Italian patients with porphyria cutanea tarda. HEPATOLOGY 1997; 27: 181-184
- Crawford DHG, Powell LW, Leggett BA, et al. Evidence that the ancestral haplotype in Australian hemochromatosis patients may be associated with a common mutation in the gene. Am J Hum Genet 1997; 57: 362-367.
- Piperno A, Arosio C, Fargion S, et al. The ancestral hemochromatosis haplotype is associated with a severe phenotype expression in Italian patients. HEPATOLOGY 1996; 24: 43-46.
- Worwood M, Dorak MT, Darke C. The ancestral haplotype for haemochromatosis- frequency of putative haemochromatosis mutations and iron overload. International Symposium on Iron in Biology and Medicine, Saint-Malo, 1997 Meeting abstract.
- Beutler E. Genetic irony beyond haemochromatosis: clinical effects of HLA-H mutations. Lancet 1997; 349: 296-297.
- Yaouanq J, Grosbois B, Jouanolle AM, et al. Haemochromatosis Cys282Tyr mutation in pyridoxine-responsive sideroblastic anaemia. Lancet 1997; 349: 1475-1476.
Copyright © 1998 by the American Association for the Study of Liver Diseases.


