
Share
Follow Us
Hepatology, May 1999, p. 1347-1351, Vol. 29, No. 5
HEPATOLOGY Concise Review
Hepatotoxicity of Psychotropic Drugs
Khaled Selim and
From the Center for Liver Diseases and the Division of Gastrointestinal and Liver Diseases, USC School of Medicine, Los Angeles, CA.
Psychotropic drugs with hepatotoxic potential can be classified based on their intended use: 1) antipsychotics-neurolepticsincluding phenothiazines, butyrophenones, and clozapine; 2) antidepressants including tricyclics, serotonin reuptake inhibitors, and monoamineoxidase (MAO) inhibitors; 3) anti-anxiety drugs such as benzodiazepines; 4) acetylcholinesterase inhibitors such as tacrine; and 5) drugs of abuse including cocaine and ecstasy. Antiseizure drugs represent another class of central nervous system (CNS) drugs, but will not be considered here. Hepatotoxicity of psychotropic drugs occurs in a variable but small proportion of users and therefore can be considered unpredictable or idiosyncratic. When these uncommon adverse events occur in association with rash, eosinophilia, and/or a rapid positive rechallenge, sufficient circumstantial evidenc eexists to ascribe the mechanism to an immune-mediated hypersensitivity reaction. Acute overt reactions to drugs tend to have clinicopathological features of hepatitis (destruction of liver parenchyma), cholestasis (impaired bile secretion), or both.
The hepatotoxic reactions to psychotropic drugs conform to these general patterns. Furthermore, as with most hepatotoxic drugs,individual psychotropic drugs have a characteristic pattern of injury, i.e., cholestatic for some (e.g., chlorpromazine, haloperidol, tricyclics), hepatitic for others (e.g., hydrazines, MAO inhibitors, cocaine, ecstasy) (see table 1).
| table 1. Hepatotoxicity Caused by Psychotropic Drugs |
NEUROLEPTICS
Phenothiazines
Phenothiazines, although no longer in widespread use, typify numerous drugs associated with liver injury. There is a highbackground of asymptomatic liver test abnormalities (>20%) and a lower incidence of overt liver disease (0.1%-1%). Features of hypersensitivity are seen in about half the phenothiazine cases(including positive rechallenge).1-4 Chlorpromazine has been the most extensively studied. The clinical features appear t obe accounted for by a mix of hypersensitivity reaction and metabolitetoxicity. The bile ductule may be an important target, and a ductopenicsyndrome is the most severe, although uncommon,consequence.
The classic description of the clinicopathological picture of phenothiazine-induced hepatic injury, written by Ishak and Ireyin 1972,3 remains the standard in the field. It describes 36 validated cases, of which 33 had received chlorpromazine. A prodromeof nonspecific symptoms lasting 1 to 2 weeks was common. Eosinophiliawas observed in three quarters. Four developed a chronic ductopenicsyndrome (over 30 have been reported in all; 3 progressed to cirrhosis,although many lacked long-term follow-up). A survey of prescriptions in the United Kingdom from 1985 to 1991 revealed an overall incidence of chlorpromazine jaundice of 0.16%, increasing to 0.3% over age70, more than 10 times higher than in those below age 50.4Cross-reactivity among phenothiazines is extremely rare,17 butavoidance of this class of compound is probably prudent in anypatient with suspected history of an overt hepatic reaction tochlorpromazine.
Most of the work on pathophysiology of experimental chlorpromazine cholestasis dates back about 20 years, implicating reactive metabolites, with damage to membranes and the cytoskeleton, andprostaglandin-induced sinusoidal perfusion abnormalities.5-8Chlorpromazine metabolism is very complex. Experimentally, itproduces a dose-related impairment in bile secretion, inhibitingNaK adenosine triphosphatase and altering membrane fluidity.9-11Ring-hydroxylated products are more potent and the sulfoxidationproduct less potent.12,13 In experimental animals, dose-relatedcholestasis is induced within minutes. Chlorpromazine is a cationicamphiphile with detergent properties; it binds to and precipitates bile acids and phospholipids. It is, however, unclear if theseeffects are responsible for cholestasis. In monkeys, phospholipidsecretion is decreased much more significantly than is bile acidsecretion.13 Although the drug’s use has declined, it wouldbe of interest to re-examine the pathogenesis of its cholestaticeffects in light of the recent advances in the knowledge of molecularmechanisms of bile secretion and drug metabolism.
Attempts to identify genetic factors in chlorpromazine hepatitis have been interesting, though limited and of uncertain significance.Because the sulfoxide seems less toxic, a genetic defect in sulfoxidationwas sought.12 The phenotyping employed S-carboxymethyl-L-cysteine,which presumably undergoes sulfoxidation as cysteine does,14-16in the pathway to sulfate formation. Thus, this test should notreflect CYP-mediated metabolism of chlorpromazine. However, all12 patients recovering from chlorpromazine jaundice were deficientin sulfoxidation.12 Could this indicate a defect in a pathwayfor detoxification of hydroxylation products (sulfation) or otherendogenous substances? Certainly, the finding that 100% were deficientin cysteine sulfoxidation compared with 22% and 23.8%, respectively,of normal and liver disease controls may provide a clue as tofactors that determine susceptibility.12 However, questionsabout the methodology for phenotyping (paper chromatography) hascast doubt on the entire thesis.15
The mechanism of phenothiazine-induced cholestatic disease remains uncertain. In favor of a hypersensitivity mechanism isthe early onset (<1 month), presence of rash and eosinophiliain some cases, and the lack of a dose-relationship in humans.However, a metabolic idiosyncratic reaction based on individual susceptibility cannot be excluded and is supported by an extensive experimental literature.
Butyrophenones
Haloperidol, while structurally similar to phenothiazines, is a very rare cause of overt liver disease. The features resemblephenothiazine-induced cholestatic injury.18 One case of ductopenicchronic cholestatic liver disease has been reported.19 Livertest abnormalities from bromperidol also have been reported.20
Other
Clozapine is an “atypical” neuroleptic; an increase in alanine transaminase (ALT), which was mild and transient, occurredin 37% of recipients.21 While this appears benign, toxic hepatitisalso has been described.22
ANTIDEPRESSANTS
Tricyclics
Most tricyclic antidepressants are potentially hepatotoxic. Amineptine, which is not used in the United States, is the mostextensively studied. Amineptine-induced liver disease is mainlycholestatic, although moderate necrosis may be seen. An immunoallergicmechanism is suggested by the occurrence of fever, rash, eosinophilia,and positive rechallenge. Amineptine is converted by microsomesinto an epoxide that is detoxified by GSH.23,24 Although poorhydroxylators are at decreased risk, 90% of whites are rapid hydroxylators(CYP2D6).23 Thus, hydroxylator status is not a useful predictorof toxicity, although it points to the role of reactive metabolitescapable of eliciting an immune response. In vitro cytotoxicitytesting indicates that lymphocytes from patients and their first-degreerelatives exhibit an increased susceptibility to killing by amineptinemetabolites, suggesting an important genetic factor. The basisfor the latter is unknown; it does not involve altered GSH orepoxide hydrase,23 although impaired detoxification presumablycould be responsible for exposure and sensitization to amineptinemetabolites. The metabolism of tianeptine is similar to that ofamineptine. The two compounds have an identical heptanoic acidside chain and, rarely, have been associated with microvesicularsteatosis.25 The side chain is metabolized by ![]() -oxidation, leadingto inhibition of medium- and short-chain fatty acid
-oxidation, leadingto inhibition of medium- and short-chain fatty acid ![]() -oxidation.26,45Thus, both drugs are converted by P450 to reactive metabolitesthat can induce a hypersensitivity reaction in genetically susceptibleindividuals. Less commonly, they induce a microvesicular steatosis;in mice, this requires much higher doses than those used therapeutically,27although one wonders if impaired oxidation of these drugs (int he presence of a competing P450 substrate or in a poor metabolizer) might lead, at least rarely, to the accumulation of sufficient levels of the parent drug to impair
-oxidation.26,45Thus, both drugs are converted by P450 to reactive metabolitesthat can induce a hypersensitivity reaction in genetically susceptibleindividuals. Less commonly, they induce a microvesicular steatosis;in mice, this requires much higher doses than those used therapeutically,27although one wonders if impaired oxidation of these drugs (int he presence of a competing P450 substrate or in a poor metabolizer) might lead, at least rarely, to the accumulation of sufficient levels of the parent drug to impair ![]() -oxidation.
-oxidation.
Imipramine can induce a cholestatic jaundice that generally is not progressive.28 Although other tricyclics (including amitriptyline,desipramine, doxepin) rarely cause liver disease, the reportedcross-reactivity should preclude their use when sensitivity toone has been suspected.29 Occasionally, hepatitis-like injuryhas been reported with tricyclics.30,31
MAO Inhibitors
MAO inhibitors, which derive from hydrazine, are all potential hepatotoxins. The experience with one, iproniazid, was disastrous:overt hepatitis occurred in 1% with case fatalities approaching20%,32 and the drug was withdrawn. Hydrazines can be metabolizedby P450 to toxic intermediates. Their metabolism and mechanismresemble that of isoniazid, also a hydrazine. One substitutedhydrazine MAO inhibitor remains available, namely phenelzine;there have been case reports of hepatitis.33
Other Antidepressants
Trazodone has been implicated as the cause of a lesion with elements of both hepatitis and cholestasis; the problem appearsto be uncommon.34-36 Hepatotoxicity from serotonin reuptake inhibitorsinhibitors such as fluoxetine and paroxetine is reported but veryrare.37,38 Nefazodone has been associated with three casesof fulminant hepatic failure within 14 to 28 weeks of startingthe drug.38a
Anti-anxiety Drugs
Benzodiazepines, such as chlordiazepoxide, diazepam, and flurazepam, have very low hepatotoxic potential, with only case reportsin the literature, usually with a cholestatic pattern.39,40
ACETYLCHOLINESTERASE INHIBITORS
Tacrine is a reversible cholinesterase inhibitor used for Alzheimer’s disease. Remarkably, in about 50% of recipients, theALT exceeds the upper limit of normal; in 25%, the value is morethan three times the upper limit, and in 2%, it is 20-fold increased.41Nearly all the toxicity is seen in the first 12 weeks. Only afew instances of jaundice have been reported. The level of eosinophiliaand the ALT are related, the toxicity is not clearly dose-related,and positive rechallenges have been described. On the other hand,ALT levels with rechallenge were lower than those associated withthe initial drug exposure. Thus, although a hypersensitivity mechanismis possible, the reaction is sufficiently atypical as to supportthe possibility of metabolic idiosyncracy. Tacrine is metabolizedby CYP1A2 to reactive metabolites that may be damaging.42 However,CYP1A2 activity, as inferred from a caffeine breath test, doesnot predict toxicity.43
Alternative hypotheses have been put forward to explain the mechanism of tacrine-induced hepatotoxicity. Tacrine is a lipophilicamine (weak base) that may exert a protonophoric effect in mitochondria,i.e., protonation in the intermembranous space with diffusioninto the matrix, followed by deprotonation and recycling of thedrug. This movement of cationic drug and its deprotonation inthe matrix depolarizes the mitochondria, resulting in decreasedadenosine triphosphate formation.44 Similar uncoupling effectshave been seen with amiodarone and perhexiline, but accumulationof the latter in mitochondria also inhibits ![]() -oxidation, leadingto fatty liver and inhibition of the respiratory chain.45 Thesetwo effects are not seen with tacrine. Another potential mechanismis based on the inhibition by tacrine of acetylcholinesterase,leading to a cholinergic coeliac ganglion-induced stimulationof an afferent sympathetic pathway, resulting in vasoconstriction,leading to impaired perfusion of the sinusoids and reperfusioninjury mediated by reactive oxygen metabolites.46 These arenot mutually exclusive hypotheses in that the former mechanismmay sensitize to the latter. Thus, tacrine undergoes high extraction,suggesting that periportal hepatocytes may take up a large proportionof the drug; the uncoupling effect would increase respirationand O2 consumption in periportal hepatocytes, thus limiting O2availability in the more distally perfused perivenular cells;superimposition of decreased O2 delivery as a result of the effecton the microcirculation would further limit O2 in the perivenularzone.
-oxidation, leadingto fatty liver and inhibition of the respiratory chain.45 Thesetwo effects are not seen with tacrine. Another potential mechanismis based on the inhibition by tacrine of acetylcholinesterase,leading to a cholinergic coeliac ganglion-induced stimulationof an afferent sympathetic pathway, resulting in vasoconstriction,leading to impaired perfusion of the sinusoids and reperfusioninjury mediated by reactive oxygen metabolites.46 These arenot mutually exclusive hypotheses in that the former mechanismmay sensitize to the latter. Thus, tacrine undergoes high extraction,suggesting that periportal hepatocytes may take up a large proportionof the drug; the uncoupling effect would increase respirationand O2 consumption in periportal hepatocytes, thus limiting O2availability in the more distally perfused perivenular cells;superimposition of decreased O2 delivery as a result of the effecton the microcirculation would further limit O2 in the perivenularzone.
The extremely high incidence of ALT elevation caused by tacrine, despite the rare occurrence of overt liver disease, was sufficientlyworrisome to lead to a very rigorous surveillance recommendationby the FDA. The concerns about hepatotoxicity and the cumbersomenature of the required surveillance (weekly ALT for 16 weeks,then monthly for 2 months, and finally every 3 months) have limitedthe use oftacrine.
DRUGS OF ABUSE
Cocaine
Cocaine hepatotoxicity has been studied experimentally in considerable detail. Toxicity is dose-related. In naive mice, coagulativenecrosis is localized to the midzonal or the centrilobular zonedepending on the strain.47-49 In mice pretreated with phenobarbital,the toxicity is increased and shifts to the periportal zone.48,49![]() -Naphthoflavone and chronic ethanol pretreatment produce sharplylocalized centrilobular damage and increased liver injury.49The presence of covalent adducts of cocaine metabolites definesthe site of injury,49 and localizes P450 mediated toxic metaboliteproduction. However, this does not necessarily prove that covalentbinding is responsible for the observedtoxicity.
-Naphthoflavone and chronic ethanol pretreatment produce sharplylocalized centrilobular damage and increased liver injury.49The presence of covalent adducts of cocaine metabolites definesthe site of injury,49 and localizes P450 mediated toxic metaboliteproduction. However, this does not necessarily prove that covalentbinding is responsible for the observedtoxicity.
Toxicity seems to depend on P450 catalyzed N-demethylation to norcocaine, which then is converted to N-hydroxynorcocaine byflavin mono-oxygenase or P450.50 The latter redox cycles tonorcocaine nitroxide by receipt of an electron from NADPH, andthe latter transfers electrons to O2, generating oxidative stress.Covalent binding of metabolites (e.g., norcocaine nitrosonium)may also be important and can be detected by immunochemical stainingof histological sections or Western blotting.51 The presenceof covalent adducts is P450-dependent and colocalizes with thezone of necrosis. It would be of interest to apply this type ofimmunohistochemistry to liver sections of patients with suspectedcocaine hepatotoxicity. Mitochondria are key targets of the oxidativestress and may further contribute to the generation of reactiveoxygen intermediates.52 An alternative route of metabolism throughhydrolysis by esterases in plasma and liver is actually the predominantroute of metabolism and generates nontoxic metabolites.48,49,53,54Esterase inhibitors potentiate hepatotoxicity by routing moreparent drug through P450 pathways. Conversely, induction of esterases(e.g. dexamethasone) prevents toxicity.54a Because cocaine isa sympathomimetic, impaired hepatic perfusion theoretically maybe a contributing factor, the presence or absence of generalizedsystemic effects notwithstanding (e.g., hypotension andhyperthermia).
Although cocaine induces oxidative stress in hepatocytes, its mechanism is controversial. As noted above, futile redox cyclingbetween N-hydroxynorcocaine and norcocaine nitroxide, consumingNADPH and generating O2 and H2O2, has been proposed. However,because the drug exhibits a type I binding spectrum to P450, onemight predict metabolic uncoupling, with cocaine causing P450to function more as an oxidase than an oxygenase,54 and thusgenerating reactive oxygenmetabolites.
Cocaine induces its own metabolism, principally by increasing expression of CYP3A.47 Norcocaine nitroxide, when administeredto mice, induces hepatotoxicity that is P450-dependent and morphologicallyidentical to that of the parent compound,55 including the factthat pretreatment of animals with phenobarbital shifts the zoneof injury from midzonal to periportal. Thus, this sheds no lighton whether the injury mechanism involves redox cycling oxidativestress or covalent binding of a toxic metabolite (Fig. 1).
|
|
Fig. 1. Oxidative metabolism of cocaine and toxicity. N-Hydroxy-norcocaine (left) can cycle with norcocaine nitroxide (middle) or undergo further metabolism by unspecified P450 to reactive alkylating species (nitrosonium ion?). Reprinted with permission.55 |
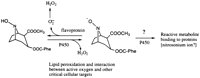
Reports in humans have documented a very small number of patients in whom cocaine seems to be an unequivocal cause of hepatotoxicity;the most convincing of these was a case with periportal necrosis.56However, most of the cases in the literature and in our clinicalexperience occur in the setting of rhadomyolysis (which itselfcan increase both aspartate aminotransferase and ALT), disseminatedintravascular coagulopathy (DIC), hypotension, hypoxemia, and/orhyperpyrexia and are associated with centrilobular necrosis whenhistology is available.53,57,58 A common additional featurehas been microvesicular steatosis in the zones spared of necrosis,57which may reflect the mitochondrial toxicity noted above. Thestudy of Silva et al. is most informative58: of 39 consecutivecases of cocaine-associated rhadomyolysis, 23 had liver abnormalities(of which the ALT in 16 was more than 10-fold increased). Hypotensionand DIC were seen in 50% of cases with liver abnormalities, andhyperpyrexia was seen in 75%. Of note, 13 of 16 cases with a markedelevation of ALT (at least 10-fold increased) developed renalfailure, whereas none of the others did, suggesting relativelysevere rhabdomyolysis and systemic effects. It should be notedthat heatstroke is a cause of hepatic necrosis, presumably asa result of impaired hepatic perfusion.59 Thus, despite thewell-documented characterization of cocaine hepatotoxicity inmice, it remains uncertain if this is more than a rarity inhumans.
Ecstasy
Ecstasy, which is 3,4-methylenedioxymethamphetamine (MDMA), produces a syndrome similar to cocaine with fulminant hyperthermia,DIC, rhabdomyolysis, and acute renal failure. Severe hepatotoxicitymay be a concomitant.60-64 MDMA is metabolized by CYP2D6; a ratmodel with deficient enzyme exhibited an elevated thermal responseto MDMA,65 suggesting that genetically poor hydroxylators (![]() 5%of whites) with decreased CYP2D6 may be predisposed to MDMA-relatedhyperthermia and possibly hepatotoxicity. However, human datato support this hypothesis arelacking.
5%of whites) with decreased CYP2D6 may be predisposed to MDMA-relatedhyperthermia and possibly hepatotoxicity. However, human datato support this hypothesis arelacking.
A number of case reports and small series describe severe acute hepatotoxicity in response to MDMA.60-64 These occurred afterincidental or regular ingestion with variable latency of daysto weeks. An eosinophilic infiltrate was seen occasionally inthe portal tracts. Repeat episodes, associated with progressivefibrosis in one case, have been described.64 Two patterns emerge:one similar to cocaine with acute profound systemic effects accompaniedby severe liver injury shortly after ingestion (hyperthermia,etc.), and the other with variably delayed, sometimes fulminanthepatitis in the absence of the systemic features and apparentlyunrelated to hyperthermia because of the latent period. MDMA willbe found in toxicology screening of the acute, but not the delayed,cases. In the latter, it is uncertain if MDMA, its metabolites,or drug contaminants are responsible. Although tissue eosinophiliais seen in some of the cases, suggesting an immune mechanism,one should bear in mind that other chemicals with direct toxicityalso are associated with eosinophilia, e.g., methylene dianiline(Epping jaundice)66 and aniline-denatured rapeseed cooking oil(toxic epidemic syndrome).67
The widespread abuse of MDMA makes it an important cause of toxic hepatitis. Because the presentation with liver injury maybe delayed and may not be accompanied by the systemic featuresof MDMA use, toxicity of the drug should be suspected in youngadults presenting with a hepatitis-like illness with negativeviral studies.68
THE USE OF PSYCHOTROPIC DRUGS IN PATIENTS WITH LIVER DISEASE
A full discussion is beyond the scope of this article. Benzodiazepines illustrate the complexities of the question: some exhibitaltered clearance in liver disease, e.g., diazepam and chlordiazepoxide,whereas others are unaffected, such as lorazepam, oxazepam, andtemazepam. Most psychotropic drugs that have been studied havedecreased clearance and increased half-life in patients with liverdisease, including midazolam, triazolam, barbiturates, tricyclics,and fluoxetine.69 However, even if hepatic metabolism is notchanged, effects of increased volume of distribution (low albuminand ascites) and increased brain sensitivity to sedation cannotbe ignored, so that dose adjustments must be made on an individualbasis. However, the low risk of hepatotoxicity with this classof drugs, coupled with a lack of evidence that underlying liverdisease would increase susceptibility to hepatotoxicity, shouldprovide reassurance that their use can follow the usual indications.The major concern is oversedation, which must beavoided.
Abbreviations
Abbreviations: MAO, monoamine oxidase; ALT, alanine aminotransferase; MDMA, 3,4-methylenedioxymethamphetamine.
FOOTNOTES
Received February 25, 1999; accepted March 12, 1999.
Address reprint requests to: Neil Kaplowitz, M.D., USC School of Medicine, 2011 Zonal Avenue, HMR 101, Los Angeles, CA 90033.
REFERENCES


